
Add comment
Comments
Hello, I found that your rationale was quite thought provoking and easy to understand! Your graphical abstract is also quite eye-catching and well laid out.
Out of curiosity I just have a quick question regarding TSPO. What potential side effects might arise from targeting TSPO in Alzheimer's treatment?
Hi, I really liked your rationale. I found it clear and easy to follow, good job on it!
I would like to know more about your proposed hypothesis - would you expect that TPSO upregulation is a causative agent in AD development or would you expect that TPSO upregulation is a sign of an already-developing AD pathology?
As you mentioned, TPSO could be an interesting target for new therapeutics. Seeing as significant upregulation is the issue, I imagine you would try to find an inhibitor-type drug. Apart from benzodiazepines and their derivatives which are known to have a neuroprotective effect against TPSO upregulation, are there any (hopefully less addictive!) drugs/ligands which could be considered as potential therapeutics?
Again, well done on the rationale and graphical abstract :)
Great question, Zoha. This is critical (whether upregulated or downregulated) as it will impact how therapeutic strategies are designed and implemented.
Hi Zoha,
Thanks for your question!
I wouldn't describe TPSO upregulation as a direct cause of Alzheimer's disease (AD), but rather as something that becomes significantly upregulated during the disease's progression.
TPSO serves as an early marker of microglial activation, and in AD, microglia can express several different "phenotypes." We expect that in the early stages of the disease, microglia will exhibit a more protective profile, while in later stages, they will shift to a more inflammatory profile. And this will be always correlated to an increase in TSPO.
It's possible that the initial increase in TSPO may reflect an attempt by the brain to repair damage. However, its upregulation may not be sufficient to prevent or slow the disease's progression.
As for therapeutic strategies and potential inhibitors, we considered PK11195, a specific TSPO antagonist, as a possible approach. However, we didn't include it in our hypothesis yet, because the mechanisms by which TSPO contributes to AD pathology are not fully understood. So, for now, we’re focusing on understanding the molecular link between TSPO and AD—and who knows, maybe we’ll accidentally uncover the perfect cure for AD along the way. ;)
Add comment
Comments
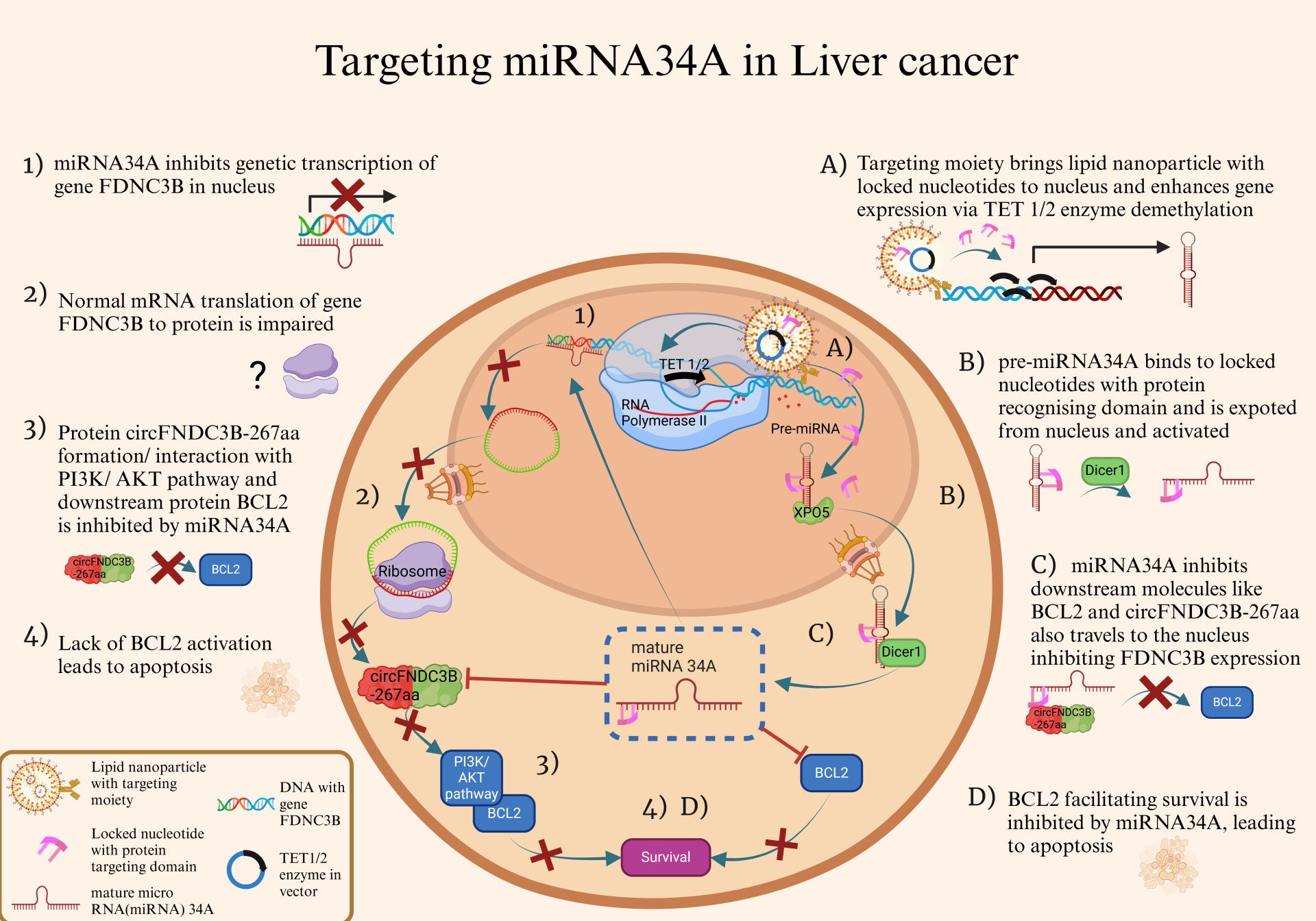
Hi, I think that your hypothesis presents an innovative approach to tackling hepatocellular carcinoma, and the graphical abstract complements it very well.
So, I have a question. Since liver tumours often have a highly immunosuppressive microenvironment, how might the upregulation of miRNA-34A interact with immune cells? Could this strategy have immunomodulatory effects that enhance or hinder anti-tumour immunity?
Great abstract very visual and easy to follow!
You mention using locked nucleotides and TET enzymes to promote inhibition of FDNC3B.
Given that TET enzymes are involved in DNA demethylation and their introduction could cause epigenetic changes beyond miRNA-34 promoter. How would you minimize off-target effects and ensure specificity ?
Great Abstract! This is a really interesting approach to tackling HCC. I'm sure miRNA must have been a difficult one to tackle!!
One thing I would be curious about is the specificity of the delivery system. I know miRNA therapies are notorious for having off-target effects. Did you guys have a look at the potential effects on unintended genes outside of miRNA34A? Likewise, would there be any unintended effects on normal liver cells?
Thank you so much Tony! We really appreciate your interest in our work. And yes, working with miRNA (especially ensuring specificity) has been quite a challenge (though interesting)!
To answer your question, we haven’t conducted a full analysis of off-target effects on unintended genes outside of miRNA-34A yet, but this is something we are considering for future research on this assignment. Our approach aims to minimise this risk by leveraging locked nucleotides to guide miRNA-34A activity toward FDNC3B, a key oncogene involved in PI3K/AKT signaling. However, we acknowledge that miRNA can have broad regulatory effects, so additional validation will be important.
As for unintended effects on normal liver cells, our strategy relies on the fact that miRNA-34A is naturally downregulated in HCC—so restoring its expression should primarily affect cancer cells rather than healthy hepatocytes. Additionally, our lipid nanoparticle delivery system is designed to enhance tumour-specific uptake, further limiting exposure to non-cancerous cells! :)
Hi Irma, thanks for your question :)
To minimize off-target effects, different methods can be used such as lipid nanoparticles or exosome-based delivery for targeted transport, and CRISPR/dCas9-TET fusion for precise demethylation. Locked nucleotides should have LNA modifications and optimized sequences for high specificity. Transient TET expression via inducible promoters or small molecules ensures controlled activity.
Hope this answers your question!
Add comment
Comments
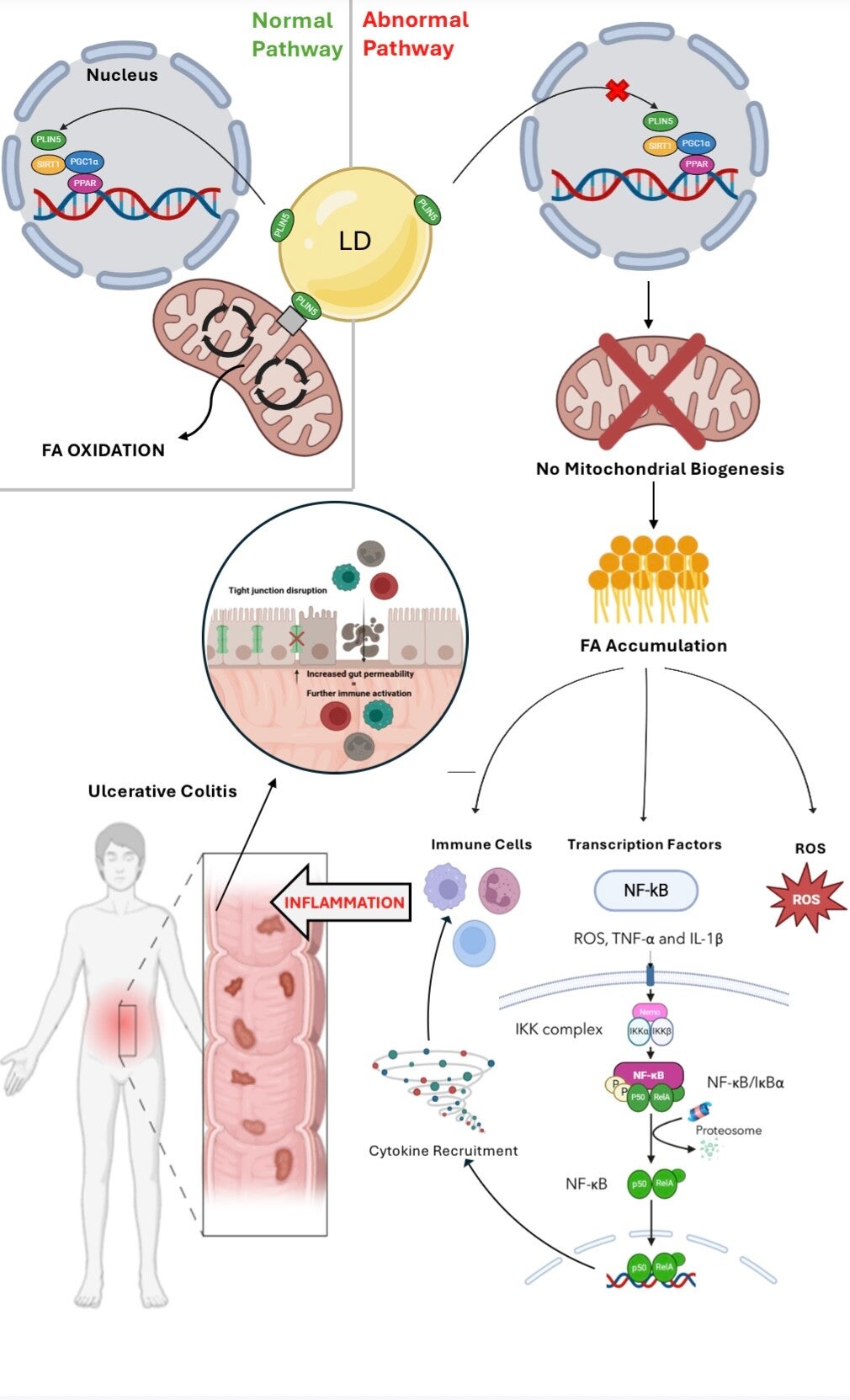
Hi all, your hypothesis sounds very intriguing! The graphical abstract helps to elucidate your theory. I just have a few thoughts to share. Considering that ulcerative colitis is characterized by periods of flare-ups followed by remission, could fluctuations in PLIN5 activity or expression levels be a factor in this pattern? What implications might this have for the timing and administration of targeted therapies? Also, since PLIN5 directly regulates lipid metabolism, its dysfunction may lead to the disturbance of bile acid synthesis. This, in turn, might alter the composition of the gut microbiota and lead to dysbiosis, another characteristic of UC. Is the interplay between altered bile acids and the composition of the gut microbiota something you have also considered? Thanks and great job once again!
Your hypothesis is really interesting, and I like how clearly you've explained it!
I was wondering, what role does oxidative stress play in linking PLIN5 dysfunction to ulcerative colitis inflammation? Does it act as the primary driver of inflammation, or are there other key factors involved in the process?
Additionally, could targeting oxidative stress directly be a potential therapeutic approach for UC, or would addressing PLIN5 dysfunction upstream be more effective?
Great job on the graphical abstract as well, it makes the concept easy to understand!
Thank you for your comment!
When PLIN5 is not functional, it cannot enter the nucleus to induce mitochondrial biogenesis. This leads to the oxidative stress, ultimately activating numerous inflammation pathways. Inflammation derived from mitochondrial dysfunction is a key factor of UC hence why the connection was made to PLIN5. Its suspected that PLIN5 is the key driver of this inflammation however the research has not been done yet so it is still unclear, of course it also is assisted by the downstream pathways like NFkB which is a key driver of inflammation.
For therapeutics, I would say that PLIN5 itself would have to be targeted to ensure effective treatment of UC. This is because if you target oxidative stress directly, the problem still remains of no mitochondrial biogenesis therefore an accumulation of FA which itself leads to inflammation in other ways like recruitment of immune cells and proinflammatory mediators.
I hope i answered your questions! let me know if you need further clarification
Hi guys, I really like your group's graphical abstract! It's very clear and easy to understand. Also, great job developing your group's hypothesis.
I just wanted to ask if dysfunction of PLIN5 solely occurs in ulcerative colitis? Or could similar lipid metabolism and oxidative stress mechanisms be involved in other gut-related inflammatory diseases like Crohn's disease or even in other inflammatory diseases in different parts of the body?
Well done again with the rationale. It seems to be a really interesting topic.
Hey Katelyn!
Thank you so much for your comment!
Yes! It is very possible that it is involved in many other inflammatory bowel diseases, or Chrons to use your example .
This would be triggered by the same pathway as described in the GA above.
It is also possible that dysfunctional PLIN5 could be associated with non fatty liver disease or many other inflammatory conditions. Much of this research is new or has not been complete yet. However, if FA accumulation can trigger NFkB pathway, it is causative of UC
I hope I answered this question, let me know if you want further clarification!
Hey, I really liked your rationale—it was very clear and easy to follow! Based on your hypothesis, what do you think would be the best approach for treating UC? Would it be more effective to focus on restoring PLIN5 function, preventing FA accumulation, boosting mitochondrial biogenesis, reducing ROS, or maybe a combination of these?
Thank you Julieann!
I think the best approach for therapies would be to focus on targeting PLIN5 function as once this is restored it will then fix downstream effects such as the ROS!
Thank you for your question :)
Add comment
Comments
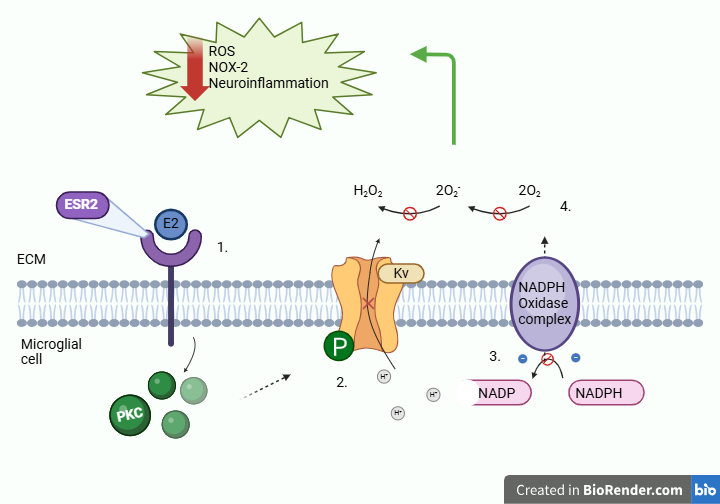
Hey guys, really liked your hypothesis and abstract especially how clearly labelled and easy to follow it was. I was just wondering about the application of ESR2 therapy. You said it could be used for Alzheimer's just wondering is there any other disorders you think it could potentially be used to treat.
Hi Darragh, we’re really glad you found the abstract clear and easy to follow. Great question! While we highlighted Alzheimer’s disease due to its strong link with neuroinflammation and oxidative stress, ESR2 modulation could potentially be beneficial for other neurodegenerative and neuroinflammatory disorders as well.
For example, Parkinson’s disease (PD) involves microglial activation and excessive ROS production, contributing to dopaminergic neuron degeneration. By reducing Kv channel activity and limiting NOX2-driven ROS production, ESR2 activation might help mitigate neuroinflammation in PD.
Similarly, Multiple Sclerosis (MS) is characterized by chronic neuroinflammation and oxidative damage. ESR2’s ability to downregulate ROS levels could help protect against inflammation-induced neuronal damage in MS.
Beyond neurodegeneration, stroke and traumatic brain injury (TBI) also involve ROS-driven secondary damage. ESR2 activation could be explored as a strategy to reduce oxidative stress and promote neuroprotection in these conditions.
Hi everyone, your graphical abstract look great and really visualizes a very intriguing hypothesis! I just have a one question for you all:
How does ESR2-mediated modulation of Kv channels influence the broader neuroinflammatory landscape, and could targeting this pathway provide a viable therapeutic strategy for neurodegenerative diseases such as Alzheimer’s? Thanks :)
Hi William, thank you for your question! Our hypothesis suggests that ESR2 activation reduces neuroinflammation by modulating Kv channel activity. As shown in our graphical abstract, estradiol (E2) binds to ESR2, triggering intracellular signaling that leads to the phosphorylation of Kv channels (Step 2). This phosphorylation alters Kv channel conductance, reducing potassium efflux and preventing excessive membrane hyperpolarization. As a result, calcium influx is limited (Step 3), which in turn decreases NADPH oxidase (NOX2) activation and lowers ROS production (Step 4).
Since ROS play a key role in amplifying neuroinflammatory pathways, reducing their production through ESR2-mediated Kv channel modulation could indeed provide a therapeutic strategy for neurodegenerative diseases such as Alzheimer's. By targeting ESR2, it may be possible to dampen microglial-mediated oxidative stress and inflammation, potentially slowing disease progression.
Hi guys, well done on the graphical abstract! It’s well laid out and really easy to follow :)
Are there other estrogen receptors, such as ESR1, that could modulate or counteract the effect of ESR2 on Kv channels in microglia? Also, could cytokines from other signalling pathways crosstalk with the ESR2 and influence ESR2’s effect on the Kv channels in microglia?
Hi Ciara, thanks for the question and your feedback! :)
The ratio of ESR1 and ESR2 expressed on microglial cells would most likely influence the response. More often ESR1 gives a pro-inflammatory response, and would upregulate calcium influx.
Selective ESR2 agonists would combat this issue.
Hi guys, I really like your graphical abstract, it’s really well laid out!
In terms of targeting ESR2 for reducing neuroinflammation, would there potentially be any off target effects?
Would the therapeutic approach be the same in males and females as estradiol, and therefore ESR2, may be expressed at different levels in males and females?
Hi Roisin,
Thanks for the feedback! As we all know ESR2 is also expressed in neurons, oligodendrocytes where it regulates synaptic plasticity and neurogenesis also in other tissues across the body. Specifically for the CNS region it could cause unintentional synaptic transmission and change neuronal plasticity. This could be combated by developing microglial-specific delivery methods or by identifying Kv channel-targeting downstream effectors.
Most of the in vivo research has been done on female mice as they have a higher ESR2 expression in the brain than males. Males are still reactive to ESR2 stimulation although not as sensitive as females. Males might need a different dose or formulation of the drug to induce the same affect.
Thank you for that question !

Add comment
Comments
Hey guys, I love this abstract! It's very clear and easy to understand the role and dysfunction of OSBPL1A in the body.
I'm just wondering, could targeting OSBPL1A-mediated lipid transport be a good therapeutic strategy for preventing α-synuclein aggregation and neuroinflammation in multiple system atrophy as the dysfunction in this mechanism causes lipid accumulation in the first place? :)
Hi guys. Well done on your great graphical abstract!
When you mention that OSBPL1A dysregulation disrupts cholesterol transport, do you think that it might also impair the ability of astrocytes to transport cholesterol to neurons? Would this then lead to a decrease in neuronal cholesterol?
:)
Hey, I really like the design of your graphical abstract, it’s really well broken down and easy to follow, great job :)
I just had a quick question about OSBPL1A dysregulation. Would there be any link between having a metabolic disorder that effects lipid metabolism, and the exacerbation of the lipid-mediated neuroinflammation caused by the dysregulation of OSBPL1A, and does this have the potential to contribute to the progression of Multiple System Atrophy in a patient ?
Thanks very much Ellen!
Yeah, conditions like diabetes or high cholesterol that affect lipid metabolism could definitely worsen OSBPL1A dysregulation and, according to our hypothesis, accelerate MSA progression. Since OSBPL1A plays a role in cholesterol transport in the brain, any extra lipid imbalance could disrupt oligodendrocytes, leading to increased α-synuclein accumulation and inflammation. Also, those metabolic disorders are known to cause chronic inflammation, which could further contribute to neuroinflammation in MSA.
Thanks again for the feedback :)
Add comment
Comments
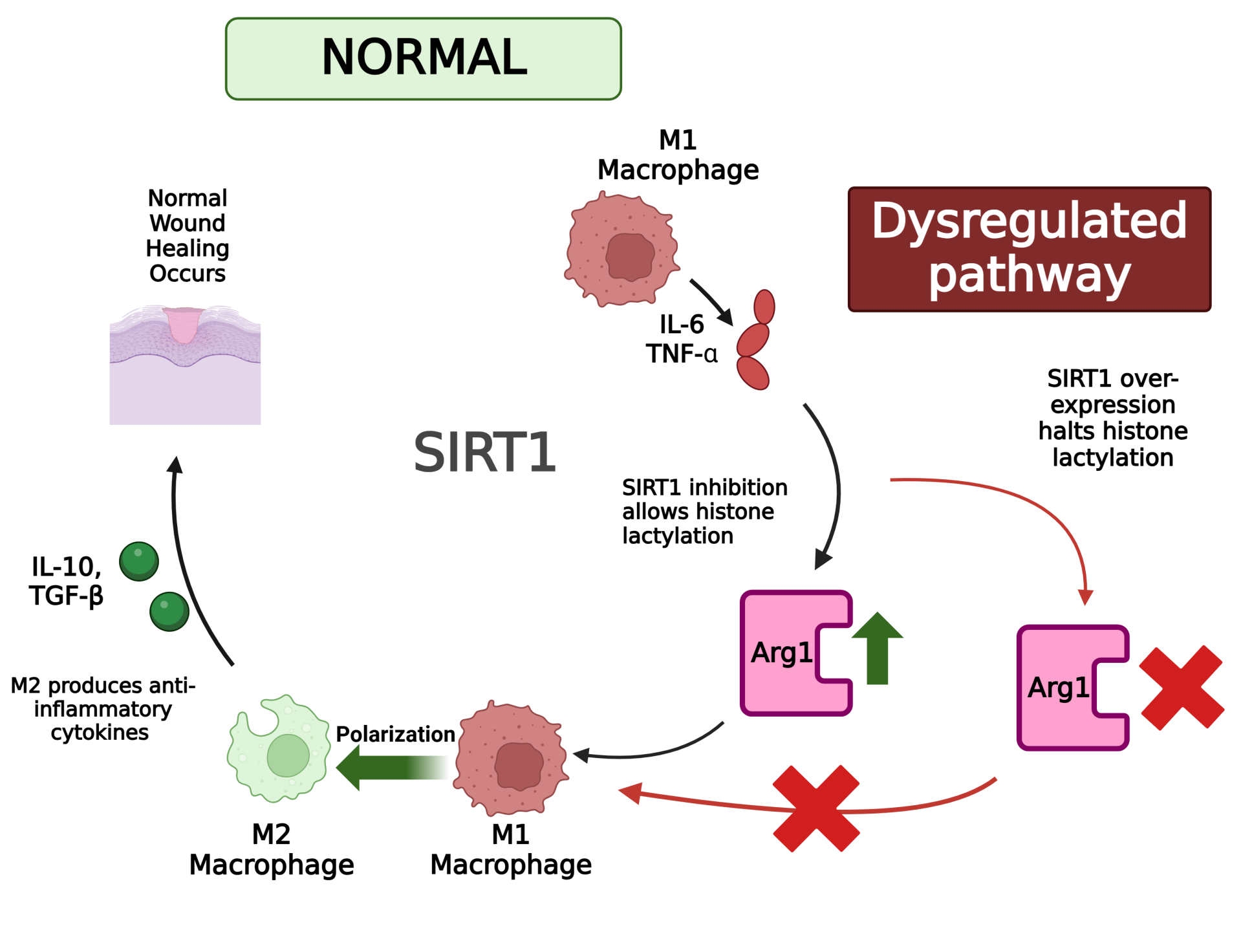
Hi guys, nice job - abstract looks great:) Was just wondering, could small-molecule inhibitors of SIRT1/histone lactylation be used to restore Arg1 expression and M2 macrophage function in chronic wounds - and would there be many challenges in targeting SIRT1 without disrupting its other homeostatic roles?
Well done on the hypothesis and rationale I found them really easy to follow, along with the graphical abstract!
I just have one question, since histone lactylation plays a role in M2 macrophage polarization, did you look into if modifying lactate metabolism rather than directly targeting SIRT1 could be a more effective strategy for enhancing wound healing?
Well done on your graphical abstract, it presents a nice clear and compelling hypothesis while also visually reinforcing the key mechanisms.
I have a question regarding the long term effects of epigenetic modifications on macrophage function, does SIRT1 over-expression induce an epigenetic memory in macrophages, enabling them resistant to M2 polarisation even after normalisation and how would this impact chronic wound healing?
Hey guys! Really good job on your graphical abstract, it follows a very clear pathway and has been presented really well.
One thing I am curious about though is the fact you mention that SIRT1 is well-known for its roles in post-translational modifications, could there be unintended consequences of inhibiting/modulating its activity in wound healing? I'd be interested in learning the specifics that SIRT1 does in general prior to the alteration.
Add comment
Comments
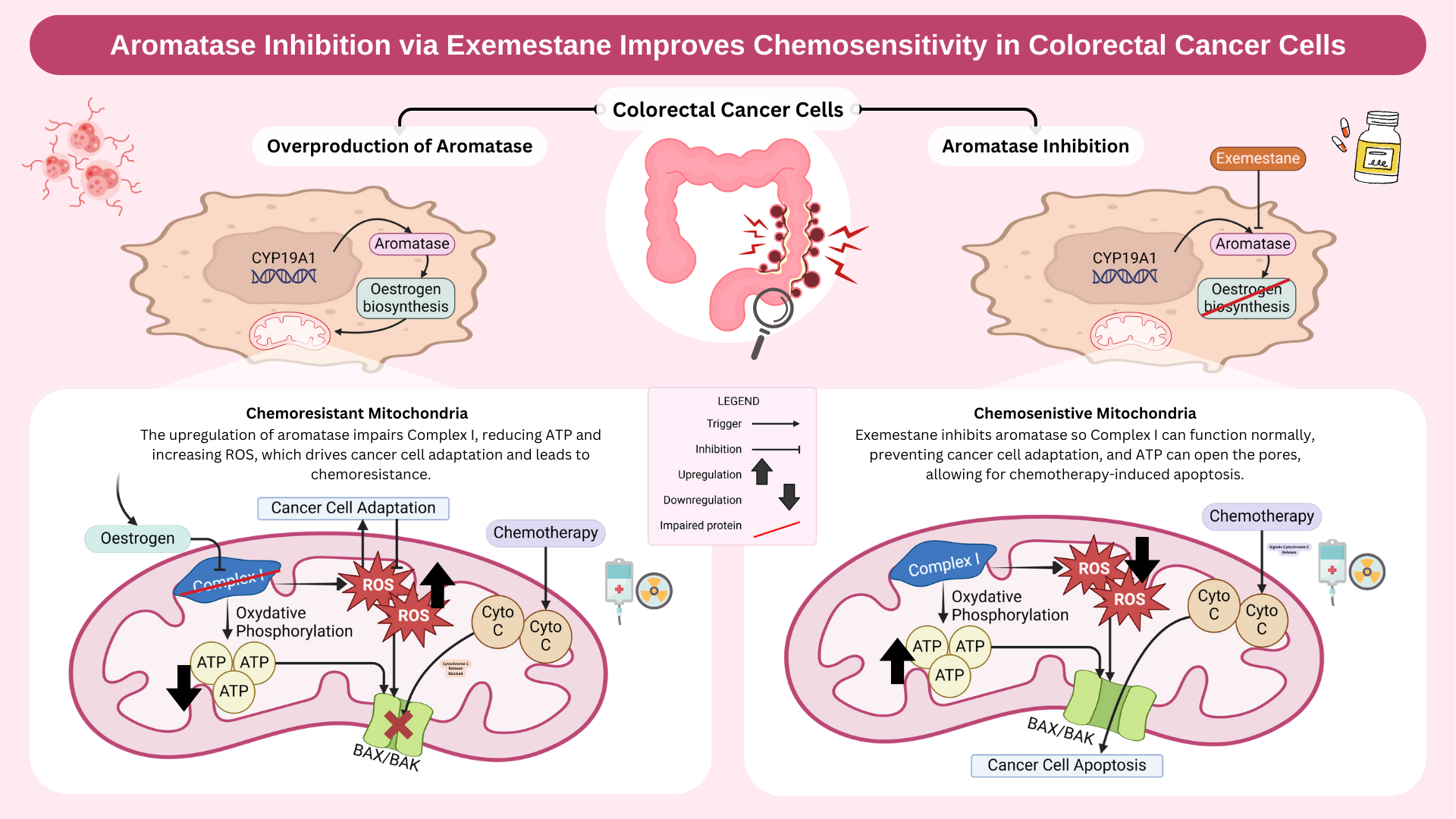
Hey guys, Lovely GA and very interesting hypothesis.
I just had a question regarding the impairment of complex 1 in the mitochondrial membrane. when this is impaired, does it have negative consequences for the other complexes in the membrane? and does this subsequently add to chemoresistance?
Hey Nyah, thanks for the question.
Yes, impairment of complex I can have cascading effects on the other complexes in the electron transport chain (ETC). Complex I plays a key role in initiating oxidative phosphorylation by transferring electrons to coenzyme Q (ubiquinone), which then passes them to complex III and IV. When complex I is impaired, it disrupts the electron flow, leading to reduced ATP production and increased ROS generation due to electron leakage. This oxidative stress can then damage other ETC complexes, particularly complex III and IV, further exacerbating mitochondrial dysfunction.
This then contributes to chemoresistance because the cancer cells adapt to the mitochondrial dysfunction using various methods such as switching to glycolysis (Warburg effect) as an alternative energy source, or upregulating antioxidant defences, making them more resistant to chemotherapy-induced oxidative stress.
I hope that answers your question!
Great question definitely one we considered! Targeting the gene (CYP19A1) would definitely have a broader longer, longer-lasting effect for sure. The issue would be achieving a tissue-specific inhibition while avoiding systemic effects!
RNA Interference (RNAi) and Gene Editing were definitely avenues of inhibition we looked into but as aromatase (CYP19A1) is expressed in so many tissues and has the fairly significant role of converting androgens into estrogens...
We’d likely see significant off-target physiological repercussions: likely disruptions to sexual development, bone health, reproductive health, and even brain function, where you see aromatase getting involved in regulating mood, cognition, and neuroprotective functions through estrogen production!
For this reason, we had a look at targeting aromatase at the protein level, hoping for a more focused approach, inhibiting the enzyme's activity and having the desired role of countering the excess estrogen biosynthesis that’s leading to the above dysfunction of complex 1.
Hi guys,
Very nicely done on the abstract! Would there be off-target adverse effects if you targeted the gene itself (CYP19A1) instead of aromatase?
That is a very interesting question. Which would cause more deleterious effects: a protein or a gene?
Hi Clara, yes it would be fair to say this strategy could still have potential off-target effects. In relation to this you could question whether exemestance is specific enough. The thinking behind this was compared to non-steroidal inhibitors such as anastrozole or letrozole, exemestance may have fewer off-target interactions, but since aromatase is expressed in the ovaries, brain etc we would likely be looking at different localized drug delivery strategies to try to bypass systemic circulation such as enteric coating or nanoparticle delivery
Add comment
Comments
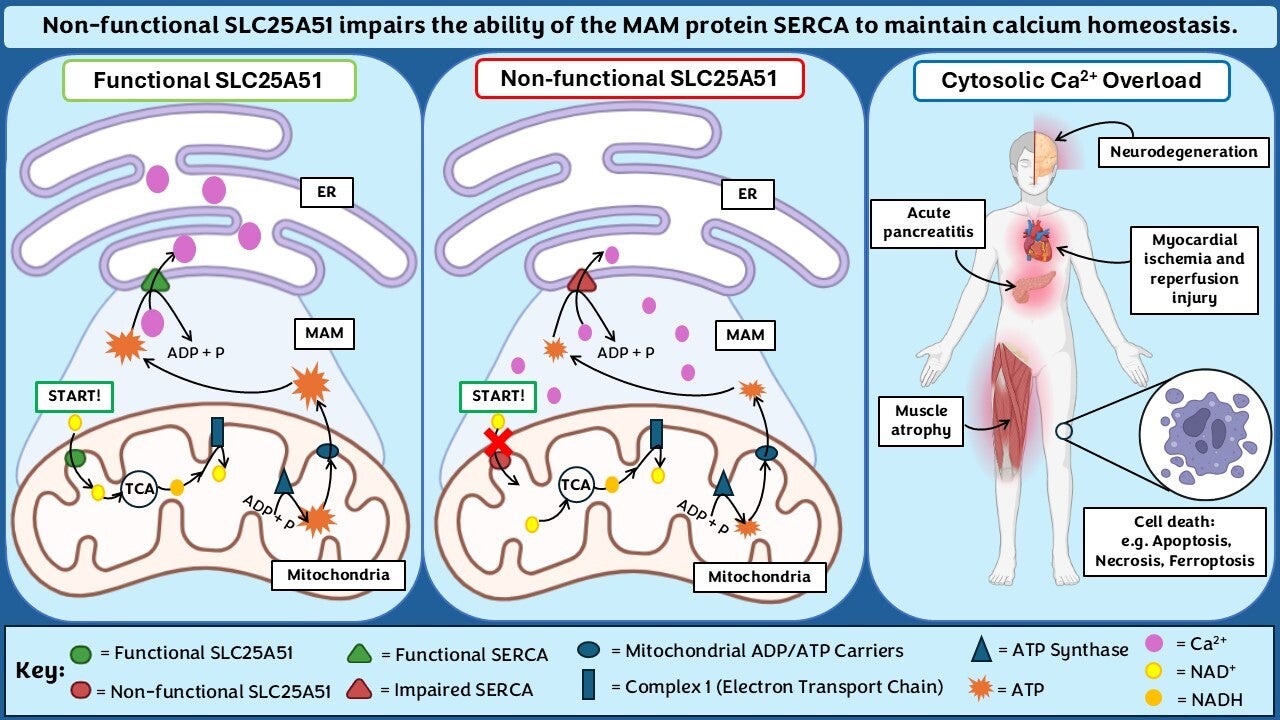
Hi guys, this is a really interesting take on the link between SLC25A51, ATP production, and calcium homeostasis! The way mitochondrial dysfunction can trigger such widespread effects, from neurodegeneration to muscle atrophy, is fascinating. It makes me wonder—could enhancing mitochondrial ADP/ATP transporters compensate for the loss of SLC25A51, or is its function too specific to be replaced?
Hi, I think that this is a compelling hypothesis that effectively links mitochondrial NAD+ transport to ATP production and calcium homeostasis. The graphical abstract also does a great job illustrating the pathway.
Does the impairment of SERCA due to ATP depletion occur uniformly across different cell types, or are certain cells more susceptible? Given that different cell types have varying ATP demands and calcium regulation mechanisms, could some tissues exhibit a greater vulnerability to SLC25A51 dysfunction than others?
Hi Catherine, thank you for your question. :)
Yes, as different cell types have varying ATP demands and calcium regulation mechanisms, this has the potential to influence their susceptibility to SLC25A51 dysfunction and the impairment of SERCA activity. As such, the impairment of SERCA activity due to ATP depletion is not likely to occur uniformly across all cell types. For example, cells with higher ATP demands or more active calcium regulation mechanisms, such as cardiac muscle cells, skeletal muscle cells, or neurons, might exhibit greater vulnerability to dysfunction in the SLC25A51 transporter, which is critical for mitochondrial NAD+ transport.
Thank you for the feedback, I hope this answers your question. :)
Hi, I really liked the hypothesis and the graphical abstract, the keys help to navigate the graphical abstract really well also.
Im have a small question: How does impaired calcium homeostasis lead to muscle atrophy?
Hi Aran, thanks for the question!
Calcium ions are needed for muscle fibre contraction. Under normal calcium homeostasis, the sarcoplasmic reticulum found in the sarcoplasm (cytosol of muscle cells) release calcium to cause muscle contraction. When calcium homeostasis becomes impaired, there will be an accumulation of calcium in the sarcoplasmic reticulum, as calcium cannot be transported. This can lead to a prolonged contraction of muscle. Continuous contraction of the muscle causes fatigue and the muscle cannot function as efficiently which leads to muscle weakness and injury and this leads to atrophy.
Also, when there is an accumulation of calcium in the cell, it can trigger the opening of the mitochondrial permeability transition pore. This may cause the release of reactive oxygen species which can also lead to muscular atrophy.
Lastly, an accumulation of calcium can induce the activation of proteolytic enzymes such as calpain which plays a role in muscle atrophy.
Thanks for the feedback, hope this answers your question.
Hi, I like how you visualise the mechanisms. Are you implying that the impairment of SERCA is due to the reduced ATP production? If that is the case, can ATP from other sources reduce the impairment of SERCA?
Also, how does the accumulation of Ca2+ in the cytosol affect the brain leading to neurodegeneration?
Hi Kaprissia, appreciate the comment! Our graphical abstract indeed suggests that impairment of the mitochondrial protein SLC25A51 leads to a subsequent deficiency in ATP production, which further hinders SERCA pumps. Since SERCA is heavily reliant on ATP, impaired mitochondrial ATP output directly affects SERCA's ability to regulate calcium levels. In theory, additional ATP could support SERCA activity. But the practicality of this depends on the local availability of ATP at the site of interest as well as the extent of mitochondrial dysfunction. If the ATP impairment is severe, it is unlikely that external sources could fully compensate. Lastly, accumulation of cytosolic Ca2+ in the brain will activate numerous enzymes and this can damage cellular structures such as membranes and DNA. In neurons, calcium dysregulation can disrupt synaptic function and over time this will contribute to neurodegenerative diseases.
Hi Manuela, thank you for your comment. For our hypothesis we are suggesting that a general reduction in the activity of SLC25A51 impairs the activity of SERCA, and therefore impacts calcium homeostasis. Hope that answers your question!
I really like your graphical abstract—great job on developing your hypothesis! I just wanted to clarify whether your hypothesis suggests that the loss of SLC25A51 function negatively impacts ATP production, or if you’re proposing that its loss leads to SERCA impairment and excessive calcium storage.
If it’s the first case, many studies have shown that reduced SLC25A51 protein levels are associated with impaired mitochondrial respiration and decreased ATP production.
Also, when you mention the loss of SLC25A51 function, are you referring to a specific mutation reported in the literature, or are you considering a general reduction in its cellular levels?
:)